Molecular Docking with AutoDock Vina
This pipeline is currently under review and construction and will be available soon at xyna.bio.
Molecular docking is a valuable tool to investigate the affinity between a ligand and a protein molecule. Imagine trying to fit a key into a lock – molecular docking is a computational technique that predicts how a "molecular key" (ligand) might fit into and bind with a "molecular lock" (protein), and how strongly they might interact.
The AutoDock Vina pipeline provides a streamlined and user-friendly way to run docking simulations. AutoDock Vina is a widely recognized and highly efficient tool, prized for its speed and accuracy in predicting the likely binding positions (also called "poses") and binding strengths between molecules.
This pipeline automates the entire docking process, taking you from initial molecular structures to interpretable results. It begins by preparing your input molecules using Meeko that converts the input files into PDBQT format. When complete, it provides you with the most probable binding poses and their estimated binding affinities (a measure of binding strength in kcal/mol). The more negative this value is, the better the interaction.
The predicted binding poses, and their associated binding energies can then be directly fed into more advanced simulations, such as the molecular dynamics, to further validate interactions and explore dynamic behaviour. This creates a powerful, closed-loop approach for investigating potential drug candidates and understanding fundamental biological mechanisms.
References:
Eberhardt, J., Santos-Martins, D., Tillack, A.F. & Forli, S. (2021). AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings. Journal of Chemical Information and Modeling.https://doi.org/10.1021/acs.jcim.1c00203 Trott, O. & Olson, A.J. (2009). AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. Journal of Computational Chemistry 31, 455-461.https://doi.org/10.1002/jcc.21334
The AutoDock pipeline is shown below. Note: It’s a simplified diagram showing the flow of data in the pipeline. It doesn’t include the precise datatypes and connections.
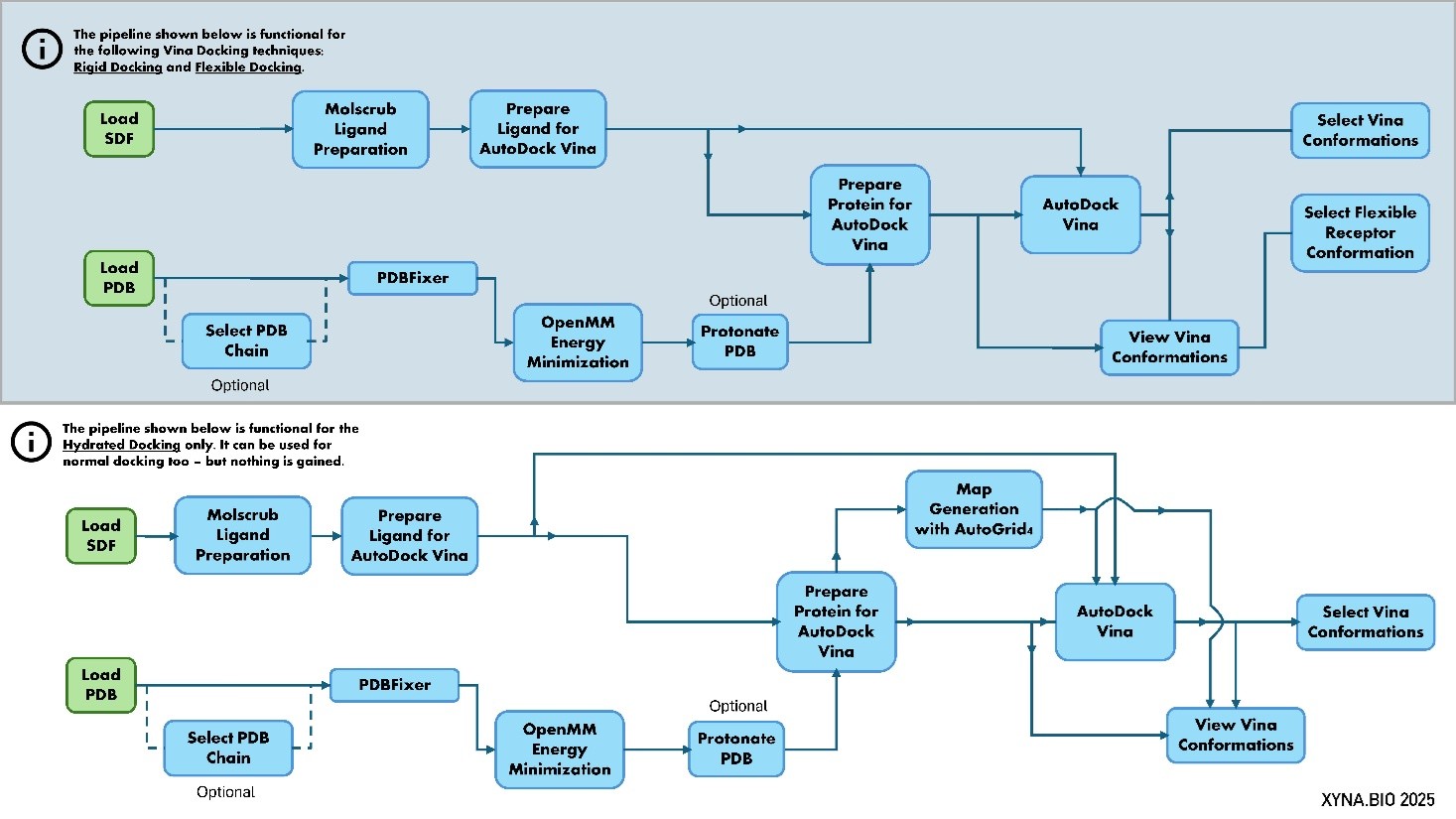
Figure 1: Schema of the AutoDock Vina pipeline. The upper version is for rigid and flexible docking, whereas the lower version is for hydrated docking. It is a simplified diagram showing the flow of data in the pipeline. It does not include the precise datatypes and connections.
1. Load Data
Use the according Load Nodes to load your data.
2. Select PDB Chain
This tool allows you to isolate and extract a specific protein chain (or multiple chains) from a larger PDB (Protein Data Bank) file. PDB files often contain multiple protein chains, or even complexes of proteins and other molecules. For many bioinformatics tasks, such as molecular docking or structural analysis, you might only need to work with a single or few of the protein chains. This node automates the process of identifying and saving only the chain(s) you are interested in, providing a clean PDB file ready for further steps.
You can choose to extract a chain by providing its specific ID (e.g., 'A, B'), or you can instruct the tool to automatically identify and extract either the largest or the smallest protein chain present in the input file based on residue count.
References:
Cock, P. J. A., Antao, T., Chang, J. T., Chapman, B. A., Cox, C. J., Dalke, A., Friedberg, I., Hamelryck, T., Kauff, F., Wilczynski, B., & de Hoon, M. J. L. (2009). Biopython: freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics, 25(11), 1422–1423.https://doi.org/10.1093/bioinformatics/btp163 Rodrigues, J. P. G. L. M., Teixeira, J. M. C., Trellet, M., & Bonvin, A. M. J. J. (2018). pdb-tools: a swiss army knife for molecular structures. F1000Research, 7, 1961.https://doi.org/10.12688/f1000research.17456.1
Input
- PDB File: The input PDB file (.pdb) from which you want to extract a specific protein chain.
Input Parameters
- Chain Selection:
- User Defined: Select this option if you know the exact ID of the chain you want to extract. You will then need to provide the PDB Chain ID.
- Largest: Automatically identifies and extracts the protein chain with the highest number of residues.
- Smallest: Automatically identifies and extracts the protein chain with the fewest number of residues.
- PDB Chain ID: A text field where you type the single letter (in all caps, e.g., 'A', 'B', 'L') corresponding to the specific chain you wish to extract. If you want multiple chains, you need to separate with a comma. E.g. 'A, E, G'. This parameter is only used if "User Defined" is selected for Chain Selection. If "Largest" or "Smallest" is chosen, this field will be ignored.
- Output File Name: A text field to specify the name of the output PDB file containing only the extracted chain. If left empty, a descriptive name will be generated automatically, typically including the original filename and the extracted chain ID (e.g., "myprotein_chain_A.pdb").
Output
- Extracted PDB File: A new PDB file (.pdb) containing only the protein chain that was selected and extracted from the input file. This file is suitable for use in subsequent steps that require a single protein chain.
3. PDBFixer
This tool is designed to "fix" common issues found in protein data bank (PDB) files, which are 3D structural descriptions of biological molecules. PDB files, especially those obtained from experimental sources, often have missing atoms or residues (building blocks of proteins), or require standardization before they can be reliably used in computational simulations.
This node uses the PDBFixer library to address these problems. It can add missing parts, replace unusual (nonstandard) residues with their common equivalents, and ensure the structure is complete and consistent. It can also add a water box around your protein and introduce ions to mimic physiological conditions. After fixing, it uses pdb-tools to renumber residues and atoms sequentially, which can be important for downstream analysis and compatibility with other software. The goal is to provide a clean, complete, and standardized protein structure ready for further processing.
References:
Eastman, P., Swails, J., Chodera, J. D., McGibbon, R. T., Zhao, Y., Beauchamp, K. A., Wang, L. P., Simmonett, A. C., Harrigan, M. P., Stern, C. D., Wiewiora, B. R., Brooks, B. R., & Pande, V. S. (2017). OpenMM 7: Rapid Development of High Performance Algorithms for Molecular Dynamics. PLOS Computational Biology, 13(7), e1005659.https://doi.org/10.1371/journal.pcbi.1005659 Rodrigues, J. P. G. L. M., Teixeira, J. M. C., Trellet, M., & Bonvin, A. M. J. J. (2018). pdb-tools: a swiss army knife for molecular structures. F1000Research, 7, 1961.https://doi.org/10.12688/f1000research.17456.1
Input
- Input PDB: The PDB file (.pdb) of the protein structure that you want to clean up and fix.
Input Parameters
- Add Missing Atoms: A dropdown menu to choose which missing atoms should be added to the PDB file.
- none: No missing atoms are added.
- heavy: Only non-hydrogen atoms (e.g., carbon, oxygen, nitrogen) are added. This is often a safe default as hydrogen positions can be ambiguous. (Default)
- hydrogen: Only missing hydrogen atoms are added.
- all: All missing atoms, both heavy and hydrogen, are added.
- Add Missing Residues: A toggle (on/off) option. If enabled, the tool will attempt to add any missing protein residues to complete the chain. Caution: This can sometimes lead to long, flexible, and potentially unrealistic loops in the structure. It is highly recommended to visually inspect the output structure if you use this option. (Default: Off)
- Replace Nonstandard Residues: A toggle (on/off) option. If enabled, the tool will identify and replace any non-standard or modified amino acid residues with their closest standard equivalents. This is useful for ensuring compatibility with other tools that expect standard protein building blocks. (Default: Off)
- Keep Heterogens: A dropdown menu to specify which non-protein molecules (heterogens, like ligands, cofactors, or water) should be kept in the fixed PDB file.
- all: Keep all heterogen molecules present in the original file.
- water: Only keep water molecules.
- none: Remove all heterogen molecules. (Default)
- Output File Name: A text field to specify the name of the new, fixed PDB file. If left empty, a name will be automatically generated, typically by appending "_fixed" to the original file name.
Expert Parameters (available via the menu icon in the node)
This node provides access to advanced parameters for both the PDBFixer and pdb-tools utilities, allowing for more specific control over the fixing process and system setup. See the full node documentation for more details.
Output:
- Output PDB: The cleaned, fixed, and sequentially renumbered PDB file. This output is a complete protein structure that is more suitable for further computational analyses and simulations.
4. OpenMM Energy Minimization
This tool optimizes the 3D structure of a protein (or other biomolecule) by performing "energy minimization" using the OpenMM library. In essence, it takes an initial structure and adjusts the positions of its atoms to find a more stable, lower-energy arrangement. This process is crucial for preparing structures for subsequent simulations like molecular docking, as it removes clashes and ensures the molecule is in a realistic, relaxed state. The node first adds any missing hydrogen atoms, then applies a chosen "force field" (a set of rules governing atomic interactions) to guide the structure towards its most stable conformation.
Reference:
Eastman, P., Swails, J., Chodera, J. D., McGibbon, R. T., Zhao, Y., Beauchamp, K. A., Wang, L. P., Simmonett, A. C., Harrigan, M. P., Stern, C. D., Wiewiora, R. P., Brooks, B. R., & Pande, V. S. (2017). OpenMM 7: Rapid Development of High Performance Algorithms for Molecular Dynamics. PLOS Computational Biology, 13(7), e1005659.https://doi.org/10.1371/journal.pcbi.1005659
Input
- Input PDB: The PDB file (.pdb) containing the 3D structure of the molecule you wish to energy minimize.
Input Parameters
- Constrain Minimization: A toggle (on/off) option. If enabled, certain parts of the molecule can be held fixed or restrained during the minimization process. When this is 'on', the "Constraint Selection" and "Constraint Stiffness" parameters in the Expert Parameters section become active. (Default: Off)
- Output Filename: A text field to specify the name of the resulting minimized PDB file. If left empty, a name will be automatically generated, typically by appending "_minimized" to the original input file name.
Expert Parameters (available via the menu icon in the node)
This node provides access to advanced parameters that give you fine-grained control over the energy minimization process. These settings allow you to define the physical environment and algorithmic details of the simulation. See the full node documentation for more details.
Output
- Minimized PDB: The 3D structure of your molecule after energy minimization, provided as a PDB file. This output represents a more stable and realistic conformation of your molecule, suitable for further computational experiments.
5. Protonate PDB
This tool prepares protein structures by adjusting their "protonation states" and adding missing hydrogen atoms based on a specified pH value. In biological systems, the charge and reactivity of amino acid residues (the building blocks of proteins) can change depending on the acidity or alkalinity (pH) of their environment. Accurately modeling these protonation states is critical for many computational simulations, such as molecular docking or molecular dynamics.
This node uses PROPKA to predict the pKa values (a measure of acidity) of titratable residues (like Aspartic acid, Glutamic acid, Histidine, Lysine, Arginine) and then employs PDB2PQR to add hydrogens and assign atomic charges according to the chosen pH and a specific "force field" (a set of parameters defining atomic interactions). This ensures your protein structure is chemically realistic for downstream simulations.
References:
Dolinsky, T. J., Czodrowski, P., Li, H., Nielsen, J. E., Jensen, J. H., Klebe, G., & Baker, N. A. (2007). PDB2PQR: expanding and upgrading automated preparation of biomolecular structures for molecular simulations. Nucleic Acids Research, 35(suppl_2), W522–W525.https://doi.org/10.1093/nar/gkm276 Jurrus, E., Engel, D., Star, K., Monson, K., Brandi, J., Felberg, L. E., Brookes, D. H., Wilson, L., Chen, J., Liles, K., Chun, M., Li, P., Gohara, D. W., Dolinsky, T., Konecny, R., Koes, D. R., Nielsen, J. E., Head-Gordon, T., Geng, W., Krasny, R., Wei, G. W., Holst, M. J., McCammon, J. A., & Baker, N. A. (2018). Improvements to the APBS biomolecular solvation software suite. Protein Science, 27(1), 112–128.https://doi.org/10.1002/pro.3280 Olsson, M. H. M., Søndergaard, C. R., Rostkowski, M., & Jensen, J. H. (2011). PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical pKa Predictions. Journal of Chemical Theory and Computation, 7(2), 525–537.https://doi.org/10.1021/ct100578z
Input
- Input PDB: The PDB file (.pdb) of the protein structure you wish to protonate.
Input Parameters:
- pH Value: A numerical value (float) representing the target pH for adjusting the protein's protonation states. The tool will protonate or deprotonate residues to match their expected state at this pH. (Range: 0.0 to 14.0, Default: 7.0)
- Force Field: A dropdown menu to select the force field that will be used. This choice influences how atomic charges and atom types are assigned during the protonation process, affecting how the protein interacts in subsequent simulations. Options include: AMBER, CHARMM, PARSE, TYL06, PEOEPB, SWANSON. (Default: AMBER)
- Output File Name: A text field to specify the desired name for the output protonated PDB file. If left empty, a descriptive name will be automatically generated, typically including the original filename and the target pH (e.g., myprotein_protonated_pH7.0.pdb).
Expert Parameters: (available via the menu icon in the node) - --drop-water: If enabled, any water molecules present in the input PDB file will be removed before the protonation process begins. (Default: False)
Output
- Output PDB: A new PDB file (.pdb) containing your protein structure with all hydrogen atoms added and the protonation states of its titratable residues adjusted to the specified pH. This file is ready for use in molecular simulations.
6. Molscrub Ligand Preparation
This tool streamlines the preparation of small molecules (ligands) for molecular docking and molecular dynamics simulations using molscrub.py. It takes an input SDF file and performs a series of cleaning and optimization steps to ensure the ligand structure is suitable for downstream computational workflows.
The preparation process can involve several advanced chemical transformations and optimizations: - pH Correction/Acid-Base Enumeration: Adjusting the protonation states of the ligand based on a target pH. - Tautomer Enumeration: Generating different structural isomers (tautomers) that are in equilibrium, focusing on low-energy states. - Ring Fixes: Correcting distorted 6-membered rings and enumerating different chair conformations (for cyclohexane-like rings). - 3D Coordinate Generation and Optimization: Creating or refining the 3D atomic coordinates of the ligand and performing energy minimization using a chosen force field to find stable conformations.
Reference:
Forli, S., & The Forli Laboratory. (2024). Molscrub: a tool for cleaning molecular files (version 0.1.1). Retrieved from https://github.com/forlilab/molscrub
Input
- Input Ligand: The ligand structure file, typically in SDF format, that you wish to clean and prepare.
Input Parameters
- Protonation pH: A numerical value (float) representing the target pH for any acid/base transformations. This ensures the ligand's protonation state is appropriate for the simulated environment. (Range: 0.0 to 14.0, Default: 7.4)
- Output Filename: A text field to specify the desired name for the output prepared SDF file. If left empty, a descriptive name will be automatically generated.
Expert Parameters (available via the menu icon in the node)
This node provides a wide range of advanced parameters for the molscrub tool. See the full node documentation for more details.
Output
- Prepared Ligand File: The resulting ligand structure file in SDF format, after all cleaning, protonation, tautomerization, and 3D optimization steps have been applied. This file is ready for use in subsequent molecular docking or molecular dynamics simulations.
7. Prepare Protein for AutoDock Vina
This tool is designed to get your protein structure ready for molecular docking simulations using AutoDock Vina. It uses the Meeko library to perform several crucial preparation steps. It converts the protein into a specialized file format called PDBQT, which AutoDock Vina understands. It also generates a vital JSON file that contains information needed for later analysis of the docking results.
A key feature of this tool is defining the "grid box," which is the 3D space around your protein where the docking simulation will occur. You can either let the tool automatically calculate this box based on your protein or a specific ligand (useful for "re-docking" a known ligand). Additionally, if your protein has parts that are known to be flexible and important for binding, you can specify these "flexible residues," allowing them to move and adapt during the docking simulation, potentially leading to more accurate predictions.
Reference:
Forli, S., & The Forli Laboratory. (2025). Meeko: interface for AutoDock (version 0.6.1). Retrieved from https://github.com/forlilab/Meeko
Input
- Receptor PDB: The 3D structure of your target protein, provided as a PDB file (.pdb). This is the protein you want to dock ligands into.
- Prepared Vina Ligand Files: : A list of PDBQT files for your ligand(s). This input is specifically used if you enable the "Gridbox from Ligand (Re-docking)" option, as it helps determine the optimal size and position of the docking grid.
Input Parameters
- Flexible Residues: A text field where you can specify amino acid residues (the building blocks of proteins) that should be treated as flexible during the docking simulation. This is defined by the protein chain ID and residue number, for example, "A:5,7,B:12" would make residue 5 and 7 on chain A, and residue 12 on chain B, flexible. If left empty, the entire protein will be treated as rigid.
- Gridbox from Ligand (Re-docking): If enabled, the docking grid box (the 3D area where the ligand will search for binding sites) will be automatically calculated based on the dimensions and position of the input ligand. This is particularly useful when you are "re-docking" a ligand back into a known binding site on a protein. If disabled, the grid box is calculated based on the entire receptor protein.
- Output Filename: A text field to specify the base name for all output files generated by this node. If left empty, a name will be automatically generated, typically based on the input protein file.
Expert Parameters (available via the menu icon in the node)
This node provides access to advanced parameters for the Meeko preparation tool, allowing for more specific control over the protein preparation and grid box definition. See the full node documentation for more details.
Output
- Prepared Vina Receptor Files: A list of files containing the processed receptor data essential for AutoDock Vina. This includes:
- A PDBQT file for the "rigid" (non-moving) part of your protein.
- A PDBQT file for the "flexible" part of your protein (if flexible residues were specified).
- A JSON file containing important metadata and mapping information needed by AutoDock Vina for subsequent analysis steps.
- Grid Parameter File (GPF): A text file (.gpf) that specifies all the parameters for the docking grid, including its center and dimensions. This file is directly used by the "Map Generation with AutoGrid4" node to create the affinity maps.
8. Prepare Ligand for AutoDock Vina
This tool is used to prepare small molecules, often called "ligands," for molecular docking simulations with AutoDock Vina. Its primary function is to convert ligand structure files (SDF) into the specific PDBQT format that AutoDock Vina requires. The PDBQT format includes not only the 3D atomic coordinates but also crucial information such as atom types and partial atomic charges, which are essential for accurate docking calculations.
The preparation process involves assigning proper atom types and charges. This node can also optionally add explicit water molecules to the ligand's structure, which is useful for "hydrated docking" simulations where water molecules in the binding site play an important role.
Reference:
Forli, S., & The Forli Laboratory. (2025). Meeko: interface for AutoDock (version 0.6.1). Retrieved from https://github.com/forlilab/Meeko
Input
- Input Ligand: The file containing the 3D structure of your small molecule (ligand) in SDF format.
Input Parameters
- Hydrate Ligand: If enabled, the tool will add explicit water molecules to the ligand structure. This is used when performing docking simulations that explicitly account for the presence and interaction of water molecules within the binding site. (Default: Off)
- Output Filename: A text field to specify the name of the output PDBQT file(s). If left empty, a name will be automatically generated, typically by appending "_prepared" to the original input file name.
Expert Parameters (available via the menu icon the node)
This node provides advanced parameters for the Meeko ligand preparation tool, allowing for fine-tuned control over how your ligand is processed. See the full node documentation for more details.
Output
- Prepared Vina Ligand Files: A list containing one or two PDBQT files for your ligand, ready to be used as input for AutoDock Vina simulations:
- The primary PDBQT file for your ligand, without explicit water molecules.
- If "Hydrate Ligand" was enabled, an additional PDBQT file for your ligand that includes explicit water molecules.
9. Map Generation with AutoGrid4
Before a small molecule (ligand) can be "docked" into a protein (receptor) to predict how they might bind, we need to prepare a 3D "map" around the protein. This map describes the protein's environment, showing where different types of atoms or chemical properties would be most favourable for interaction.
This node uses a specialized tool called AutoGrid4 to create these essential "affinity maps." It takes a setup file (called a Grid Parameter File, or GPF) that defines the precise region around the protein to be mapped. The output includes several grid map files (one for each atom type or interaction), which other molecular docking tools, like AutoDock Vina, then use to efficiently calculate how well a ligand might fit and bind. It also generates a log file summarizing the mapping process. Optionally, it can also create a "water map" if your GPF is configured to include the effect of water molecules in the binding site.
References:
Forli, S., & Olson, A. J. (2012). A force field with discrete displaceable waters and desolvation entropy for hydrated ligand docking. Journal of Medicinal Chemistry, 55(2), 623–638.https://doi.org/10.1021/jm2005145
Input
- Prepared Vina Receptor Files: This input typically comes from a "Prepare Protein" node. It includes the 3D structure of your protein in a special format (PDBQT files) and any associated supporting information.
- Grid Parameter File (GPF): A text file (.gpf) that contains all the settings for AutoGrid4, such as the dimensions and location of the 3D grid, and which atom types to calculate affinities for. This file is usually generated during the initial protein preparation steps.
Input Parameters
- Generate Water Map Files: If set to 'on', AutoGrid4 will also generate an additional map file specifically accounting for the potential presence of water molecules in the binding site. This can sometimes improve the accuracy of docking predictions for certain systems. (Default: Off)
Output
- AutoGrid Map Files: A ZIP-file of generated 3D grid files (.map files). Each file in this ZIP-file represents a different type of interaction or atom affinity within the grid. These maps are crucial inputs for subsequent molecular docking simulations.
- Output GLG: The Grid Log File (.glg). This file contains a detailed record of the AutoGrid4 process, including messages about its progress, any warnings, and confirmation of successful completion. It can be useful for troubleshooting or verifying that the mapping process ran as expected.
10. AutoDock Vina
This powerful tool performs molecular docking, a computational method used to predict how a small molecule (ligand) binds to a larger molecule (protein or receptor). It simulates the process of molecular recognition, searching for the most favorable orientations and positions of the ligand in a protein.
Important Note: This tool is generally not suitable for "metalloproteins" (proteins that contain metal ions as part of their structure), as it may lead to inaccurate results.
After the simulation, the node will display the top three best binding results directly. It also generates a comprehensive file containing all predicted binding conformations of the ligand, which can be further explored using the View Vina Conformations, Select Vina Conformation, or Add SDF to PDB nodes for detailed visualization of ligand-protein interactions.
References:
Eberhardt, J., Santos-Martins, D., Tillack, A. F., & Forli, S. (2021). AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings. Journal of Chemical Information and Modeling.https://doi.org/10.1021/acs.jcim.1c00203 O'Boyle, N. M., Banck, M., James, C. A., Morley, C., Vandermeersch, T., & Hutchison, G. R. (2011). Open Babel: An Open Chemical Toolbox. Journal of Cheminformatics, 3, 33.https://doi.org/10.1186/1758-2946-3-33 Trott, O., & Olson, A. J. (2010). AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. Journal of Computational Chemistry, 31(2), 455–461.https://doi.org/10.1002/jcc.21334
Input
- Prepared Vina Receptor Files: This input comes from the "Prepare Protein for AutoDock Vina" node. It includes the protein structure (rigid PDBQT), potentially a flexible part (flexible PDBQT), and a JSON file containing essential information about the receptor.
- Prepared Vina Ligand Files: This input comes from the "Prepare Ligand for AutoDock Vina" node. It is a list containing the prepared PDBQT file(s) for your ligand, which may include both non-hydrated and hydrated versions depending on your previous settings.
- AutoGrid Map Files: (Optional) This input is a list of grid map files generated by the "Map Generation with AutoGrid4" node. It is only required if you intend to perform "hydrated docking," which considers the role of water molecules in the binding process. _ Grid Parameter File (GPF): This file contains the precise coordinates and dimensions of the 3D grid box, defining the search space for the ligand around the protein. It is typically generated during the receptor preparation step.
Input Parameters
- Exhaustiveness: An integer value that controls the computational effort AutoDock Vina spends searching for binding poses. Higher values (e.g., 32 or 64) lead to a more thorough search and generally more consistent and accurate results, but require more computation time. Lower values (e.g., 8) are faster but might miss optimal binding modes. (Range: 1 to 512, Default: 8)
- Target Number of Conformations: An integer value specifying how many different binding poses (conformations) of the ligand you want AutoDock Vina to report. While you request a number, it's important to note that the tool might not always reach this exact number if fewer distinct poses are found. (Range: 3 to 200, Default: 5)
- Output File Name: A text field to define the base name for all the files generated by this docking simulation. If left empty, a name will be automatically created based on the input receptor and ligand names.
Expert Parameters (available via the menu icon in the node)
This node provides access to advanced parameters for both AutoDock Vina and OpenBabel, offering fine-tuned control over the docking simulation and subsequent file processing. See the full node documentation for more details.
Output
- Docking Conformation 1: An SDF file representing the ligand's predicted binding pose with the highest affinity (most favorable binding).
- kcal/mol | Binding Energy for Conformation 1: The calculated binding energy (in kilocalories per mole, kcal/mol) for the highest affinity conformation. A more negative value indicates stronger binding.
- Docking Conformation 2: An SDF file for the ligand's second highest affinity binding conformation.
- kcal/mol | Binding Energy for Conformation 2: The calculated binding energy for the second highest affinity conformation.
- Docking Conformation 3: An SDF file for the ligand's third highest affinity binding conformation.
- kcal/mol | Binding Energy for Conformation 3: The calculated binding energy for the third highest affinity conformation.
- All Ligand Conformations: A PDBQT file containing all the ligand conformations predicted by the simulation. This file is useful for detailed analysis and visualization of all possible binding poses.
- All Docking Metrics: A CSV (Comma Separated Values) file containing a table of all predicted binding affinities and RMSD (Root Mean Square Deviation) values for each conformation generated during the docking simulation. This provides a comprehensive overview of the results.
11. View Vina Conformations
This tool helps you visualize the results of your molecular docking simulations performed with AutoDock Vina. After docking, Vina generates a file containing multiple possible ways (conformations) your small molecule (ligand) could bind to the protein. This node takes these results and creates a single "movie-like" PDB file that shows each predicted ligand binding pose alongside the protein receptor. If your docking simulation involved flexible parts of the protein, this tool will also correctly update their positions for each ligand pose. The resulting file is ideal for viewing in molecular visualization software like Mol* to analyze the different binding modes.
Additionally, for advanced hydrated docking simulations, this node can process the results to score the contributions of water molecules involved in the ligand-protein interactions.
Reference:
Forli, S., & The Forli Laboratory. (2025). Meeko: interface for AutoDock (version 0.6.1). Retrieved from https://github.com/forlilab/Meeko
Input
- Prepared Vina Receptor Files: This input typically comes from the Prepare Protein for AutoDock Vina node. It includes the rigid and flexible PDBQT files of your receptor and a crucial JSON file that contains mapping information necessary for accurately processing the docking results.
- All Ligand Conformations: The PDBQT file output directly from the AutoDock Vina docking process. This file contains all the predicted binding poses (conformations) of your ligand.
Input Parameters
- Score Hydrated Docking Results: If enabled, the tool will analyze the contribution of water molecules to the docking results using a specialized script (dry.py). This option requires a "Water Map File" from the AutoGrid4 mapping step. (Default: Off)
- AutoGrid Map Files: A list of grid map files generated by the Map Generation with AutoGrid4 node. This input is only required if you enable "Score Hydrated Docking Results", as it provides the water map needed for the analysis.
- Output File Name: A text field to specify the name of the combined multi-MODEL PDB file. If left empty, a descriptive name will be automatically generated.
Output
- Merged Protein Ligand Movie: A multi-MODEL PDB file (.pdb). This file contains the receptor structure, and for each predicted ligand pose, it includes the ligand's coordinates placed within the receptor. If flexible residues were defined in the receptor preparation, their positions will also be updated for each pose. This single file can be loaded into molecular visualization software to play an animation or sequentially view all predicted binding conformations. If "Score Hydrated Docking Results" was enabled, the first model in the movie will also include the scored conserved water molecules.
12. Select Vina Conformation
This tool helps you examine and extract specific results from molecular docking simulations performed with AutoDock Vina. After a docking run, AutoDock Vina generates a file containing multiple possible ways (conformations) a small molecule (ligand) could bind to a protein. This node allows you to select one of these predicted binding poses and save it as a separate file for further analysis or visualization. This is particularly useful for focusing on the most promising binding pose or comparing different poses.
Reference:
O'Boyle, N. M., Banck, M., James, C. A., Morley, C., Vandermeersch, T., & Hutchison, G. R. (2011). Open Babel: An Open Chemical Toolbox. Journal of Cheminformatics, 3, 33.https://doi.org/10.1186/1758-2946-3-33
Input
- All Ligand Conformations: This is the output file (in PDBQT format) directly generated by the AutoDock Vina tool. It contains a collection of all the different predicted binding poses (conformations) of your ligand within the protein's binding site, along with their associated binding energies.
Input Parameters
- Model Number: An integer that specifies which particular binding pose (conformation) you want to extract from the AutoDock Vina output. AutoDock Vina typically ranks its predictions, with Model 1 usually being the most favorable (lowest binding energy). You would input '1' to select the top-ranked model, '2' for the second-ranked, and so on. The numbering starts from 1. (Default: 1)
- Output File Name: A text field to specify the name of the new file that will contain only the chosen conformation. The output file will be saved in SDF format. If you leave this field empty, a descriptive name will be generated automatically based on the input file and the selected model number.
Output
- Ligand Structure File: An SDF (Structure-Data File) file containing the 3D coordinates and structural information of the single, chosen ligand conformation. This file can be used with molecular visualization software to inspect the binding pose or as input for other downstream analysis tools.
13. Select Flexible Receptor Conformation
This tool helps you examine and extract specific receptor conformations from molecular docking simulations performed with AutoDock Vina, especially when flexible residues were defined during the protein preparation. After a docking run, particularly one involving flexible receptor components, AutoDock Vina (via the "View Vina Conformations" node) generates a multi-MODEL PDB file where each model represents a predicted ligand binding pose, and the flexible parts of the receptor are also updated for each pose. This node allows you to select one of these predicted receptor conformations (corresponding to a specific ligand pose) and save it as a separate PDB file for further analysis or visualization. This is particularly useful for focusing on the receptor's induced fit or conformational changes in response to ligand binding.
Input
- Merged Protein-Ligand 3D File: The input PDB file (.pdb) that contains the multi-MODEL conformations of the ligand with the receptor. This file is generated by the View Vina Conformations node.
Input Parameters
- Receptor Model Number: An integer that specifies which particular receptor conformation (model) you want to extract from the input multi-MODEL PDB file. AutoDock Vina typically ranks its predictions, with Model 1 usually being the most favorable (lowest binding energy). You would input '1' to select the top-ranked model, '2' for the second-ranked, and so on. The numbering starts from 1. (Default: 1)
- Output File Name: A text field to specify the name of the new file that will contain only the chosen receptor conformation. The output file will be saved in PDB format. If you leave this field empty, a descriptive name will be generated automatically based on the input file and the selected model number.
Output
- Receptor Structure File: A PDB file containing the 3D coordinates and structural information of the single, chosen receptor conformation. This file can be used with molecular visualization software to inspect the receptor's conformation corresponding to a specific ligand binding pose, or as input for other downstream analysis tools like GROMACS or docking etc.
Helper Tool: OpenBabel Converter
This tool provides a versatile interface to OpenBabel, a powerful and widely used open-source toolkit for chemical information. It primarily functions as a chemical file format converter, allowing you to transform molecular structures from one file format (like SMILES or PDB) into another (like SDF or PDBQT). Beyond simple conversion, this node can also perform various chemical data manipulations, such as adding molecular properties, generating 2D or 3D atomic coordinates, and adjusting partial charges. This makes it an indispensable tool for preparing and manipulating chemical data for a wide range of bioinformatics and cheminformatics workflows.
Reference:
O'Boyle, N. M., Banck, M., James, C. A., Morley, C., Vandermeersch, T., & Hutchison, G. R. (2011). Open Babel: An Open Chemical Toolbox. Journal of Cheminformatics, 3, 33.https://doi.org/10.1186/1758-2946-3-33
Input
- Input File: A dropdown list where you select the chemical structure file from your workspace that you want to process with OpenBabel.
Input Parameters
- Input File Format: A dropdown list to specify the format of your selected input file (e.g., smi for SMILES, pdb for Protein Data Bank). It's crucial to select the correct format for accurate processing.
- Output File Format: A dropdown list to choose the desired format for the output file (e.g., sdf for Structure-Data File, pdbqt for AutoDock PDBQT).
- Output File Name: A text field to specify the name of the converted output file. If this field is left empty, the tool will automatically generate a name based on the input file's name, appending "_converted" and the new file extension.
Expert Parameters (available via the menu icon in the node)
This node exposes a range of advanced options for the OpenBabel tool, allowing for fine-grained control over the conversion and manipulation process
Output
- Output File: The resulting chemical file, converted into the specified output format and incorporating any modifications made by the chosen parameters. The exact type of the output file will match the Output File Format you selected. See the full node documentation for more details.